最近发布

高被引华东师大汤静教授联合昆士兰大学Yamauchi教授利用RapidXAFS揭秘:CO₂转化多碳选择性新纪录

全文速览
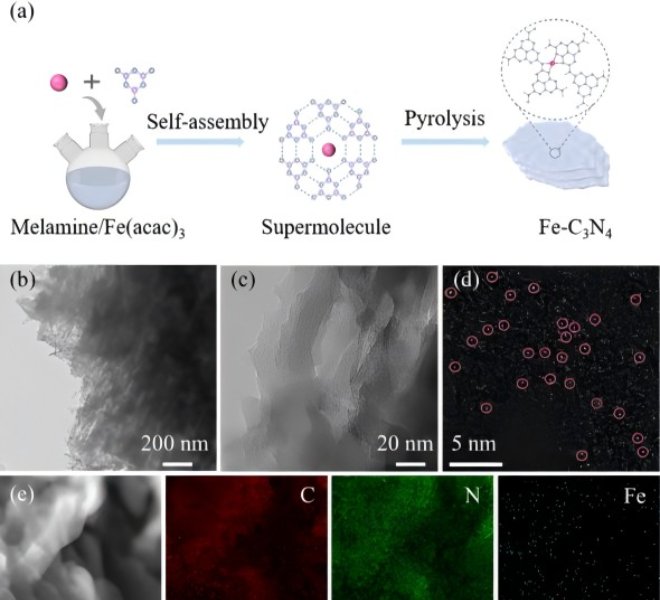
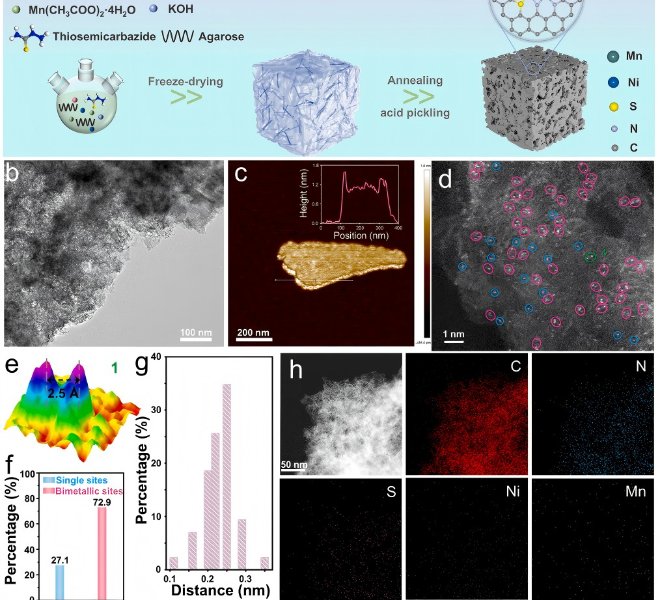
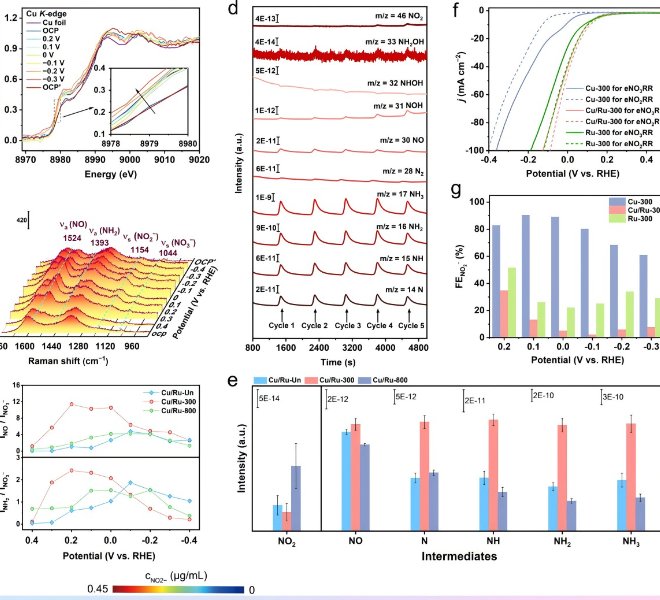
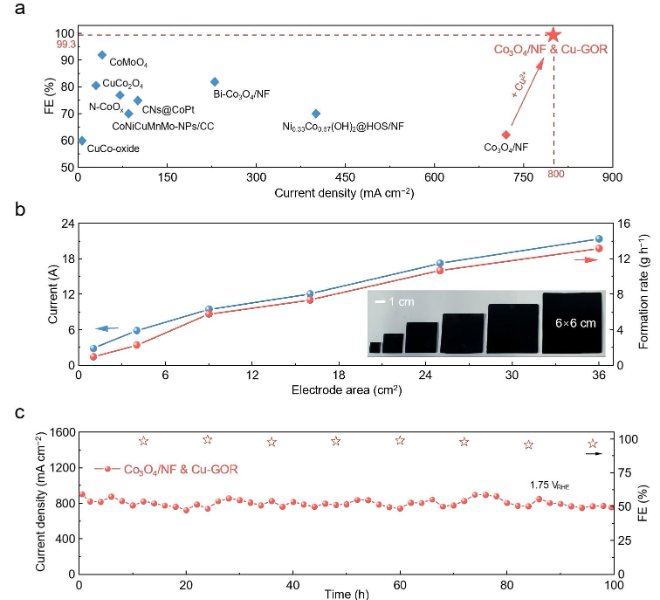
随着“双碳”目标的持续推进,如何将温室气体二氧化碳(CO₂)转化为高附加值的燃料和化学品,已成为绿色化学与能源领域的前沿焦点。近日,华东师范大学化学与分子工程学院汤静研究员与日本早稻田大学/澳大利亚昆士兰大学的Yusuke Yamauchi教授合作,在CO₂电还原(ECR)领域取得重要突破!研究团队在Angewandte Chemie International Edition上发表题为“Behavior Regulation of *CO over Self-Evolution Tandem Catalysts Under Tuned Interfacial Electric Field Boosts CO2 Electroreduction ”的研究论文。该工作系统研究了串联催化剂体系(Cu, Ag/Cu, Pd/Cu, Au/Cu)*CO中间体在不同界面电场强度下的吸附能力,并成功设计合成了三种自进化串联催化剂。研究团队关键性地采用了安徽吸收谱仪器设备有限公司推出的高性能台式X射线吸收精细结构谱仪(RapidXAFS 2M),捕捉到了催化剂在真实反应环境下的动态结构信息,解决了传统表征手段‘看得见静态,看不清动态’的痛点,为理性设计催化剂提供了坚实的数据基石。结果表明,Ag/Cu催化剂凭借适中的*CO吸附能力和低反应能垒,在-1.8 V(vs. RHE)下实现C₂₊法拉第效率高达89.2%,电流密度达553.9 mA cm⁻²,创下该类催化剂的新纪录。 电催化CO₂还原(ECR)技术是实现“双碳”目标的关键路径之一,其核心挑战在于如何高选择性地将CO₂转化为高附加值多碳产物(C₂₊)。Cu基催化剂虽潜力巨大,但CO中间体覆盖度低、C–C耦合困难等问题制约其发展。串联元素复合、自进化缺陷工程以及界面电场调控是提高CO覆盖度的有效策略,然而其协同机制与结构–性能关系仍不明确。深入理解催化剂的电子结构动态变化,必须依托于高精度、高时间分辨的原位表征技术,如X射线吸收光谱(XAS)。 本文亮点 01 通过DFT计算揭示了串联催化体系中(Ag/Cu, Pd/Cu, Au/Cu),在不同界面电场下对CO的吸附能变化规律,发现增强电场可显著强化CO吸附。 02 结合安徽吸收谱仪器设备有限公司RapidXAFS 2M提供的原位XAFS数据,证实了催化剂在电还原过程中结构稳定、配位数基本不变,为串联催化机制提供了直接结构证据。 03 明确*CO吸附能力(而非迁移能力)是决定C₂₊选择性的主导因素,Ag/Cu因其适中的吸附强度和低能垒表现最佳。 图1. 串联催化剂中*CO吸附行为的理论计算 通过DFT构建Cu、Ag/Cu、Pd/Cu和Au/Cu模型,系统计算不同电场强度下CO的吸附能。结果明确显示,随着界面电场增强,CO在各类催化剂表面的吸附能力普遍提升,其中Au/Cu因吸附能力最弱导致*CO易脱附,不利于C–C耦合。 图2. 自进化串联催化剂的制备与多尺度表征 采用水热法与电化学还原法制备M/CuO前驱体及M/Cu目标催化剂,通过XRD、SEM、TEM、XPS等手段验证其结构成功构建。尤其依托安徽吸收谱仪器公司RapidXAFS开展EXAFS分析,表明引入Ag、Pd、Au并未改变Cu的局域配位环境,M/CuO中Cu–O及Cu–Cu键配位数与纯CuO一致。 图3. 催化剂自进化过程及电催化性能 利用原位XRD追踪CV活化过程中CuO至Cu的相变过程,证实催化剂在反应中结构稳定。通过安徽吸收谱仪器公司RapidXAFS 2M采集的原位XANES数据进一步表明,Cu及M/Cu在电还原过程中配位数保持在10.6–11.3之间,说明自进化过程结构稳健,串联元素未影响Cu活性位点的电子结构。 图4. 电化学性能与*CO迁移机制的实验与模拟研究 Ag/Cu在-1.8 V下实现C₂₊法拉第效率89.2%,电流密度553.9 mA cm⁻²,显著优于其他催化剂。COMSOL模拟表明,在纳米尺度下外电场对CO“外部迁移”的影响可忽略;DFT计算则显示内迁移能垒随电场增强而升高,但CO吸附增强仍起主导作用。 图5. 反应路径与机理验证 原位拉曼光谱显示,随着电位变负(电场增强),CO吸附信号逐渐增强,与DFT预测一致。吉布斯自由能计算表明,串联元素复合有效降低了反应能垒,其中Ag/Cu能垒最低、C₂₊路径最优。结合原位红外与拉曼结果,全面验证了界面电场CO吸附C₂₊选择性的构效关系。 该研究通过DFT计算构建了四种在不同界面电场强度下的CO吸附模型(Cu、Ag/Cu、Pd/Cu和Au/Cu)。结果表明,随着界面电场强度的增加,CO吸附能力在一定程度上增强。基于此,研究人员设计了三种自进化串联催化剂(Ag/Cu、Pd/Cu和Au/Cu催化剂),借助安徽吸收谱仪器公司先进的RapidXAFS技术实现了对催化剂局域电子结构的精准解析,并在恒电位下进行了ECR测试。Pd/Cu的低C₂₊选择性归因于其高反应能垒和低催化活性。Au/Cu的高CO选择性归因于其对CO的弱吸附能力,这导致生成的CO逃逸。Ag/Cu催化剂凭借低反应能垒和适中的*CO吸附能力,在-1.8 V(vs. RHE)下实现了89.2%的高C₂₊法拉第效率(FEc₂₊)和553.9 mA cm⁻²的分电流密度(jc₂₊)。 COMSOL多物理场模拟表明,在纳米尺度上,电场对CO外部迁移的影响几乎可以忽略不计。DFT计算和实验结果均表明,与CO的内部迁移相比,CO的吸附能力主导了串联催化剂中通过C-C耦合生成多碳产物。原位ATR-IR和原位拉曼光谱实验关于CO与界面电场强度之间关系的结果与计算结果一致。该工作为研究串联催化剂中界面电场下的*CO吸附能力和迁移提供了新思路,也为多组分串联催化剂的设计提供了见解。 其这项工作为高效燃料电池催化剂的设计提供了新的思路和方向,是‘双碳’目标下碳资源循环利用技术的重大进展,也突显了国产高端表征仪器——安徽吸收谱仪器设备有限公司的RapidXAFS在前沿催化研究中的关键支撑作用。背景介绍
图文解析





总结与展望